Von Hippel-Lidau (VHL) sindromas yra santykinai reta būklė, kurios dažnis yra 1 iš 35000 gimusiųjų per metus. Liga pavadinta vokiečių gydytojo von Hippel ir švedo Lindau garbei, kurie pirmieji 1926 m. aprašė šią būklę. VHL būdingas padidėjęs kraujagyslių vešėjimas, kurios formuojasi įvairiuose organuose. Šie nenormaliai vešančių kraujagyslių dariniai gali patys sukelti problemų arba spausti aplinkui esančius organus, todėl labai svarbu, kad pacientus nuolat stebėtų patyrusi gydytojų komanda (onkogenetikas, akių gydytojas, endokrinologas, chirurgas, neurologas, pediatras, psichologas).
Kas yra von Hippel-Lindau sindromas?
Kokie yra von Hippel-Lindau sindromo požymiai?
Kaip paveldimas von Hippel-Lindau sindromas?
Onkogenetinis VHL geno tyrimas
Stebėsena esant von Hippel-Lindau sindromui
Kas yra von Hippel-Lindau sindromas?
Kokie yra von Hippel-Lindau sindromo požymiai?
Pagrindinės vietos, kuriose gali pasireikšti VHL būdingi požymiai:

Von Hippel-Lindau sindromas
VHL sergantys asmenys turi padidėjusį polinkį šiems požymiams:
| Organai |
Požymis |
Pasireiškimo dažnis |
| Akys | Tinklainės hemangioblastoma | 25-60 proc. |
| CNS | Hemangioblastomos: | |
| smegenėlių | 40-70 proc. | |
| nugaros smegenų | 13-50 proc. | |
| Inkstai | inkstų cistos | iki 45 proc. |
| šviesių ląstelių karcinoma | 25-40 proc. | |
| Antinksčiai | feochromocitoma (šerdinės dalies navikas) | 10-20 proc. |
| Kasa | kasos cistos | 40 proc. |
| kasos navikai | 5-10 proc. |
Klinikinė VHL diagnozė gali būti nustatyta, jei asmeniui yra du ar daugiau VHL būdingi požymiai ir bent vienas šių požymių yra tinklainės ar CNS hemangioblastoma. Hemangioblastoma yra gėrybinis nervų sistemos kraujagyslių ląstelių pirmtakų navikas, kuris gali sukelti vidinį smegeninį kraujavimą ir todėl turi būti nuolat stebimas. CNS hemangioblastomos paprastai šalinamos chirurgiškai, kai yra simptomatika. Kartais galima taikyti neinvazyvų gydymą gamaspinduliais (gama-skalpelis).
Akių hemangioblastomos dažniausiai nesukelia jokių požymių, tačiau gali būti aklumo priežastimi, todėl reikia nuolat stebėti akių dugną ir šalinti angiomas ankstyvose stadijose (lazerio arba krio-terapija). Ankstyva diagnostika palengvina gydymą ir padeda išvengti komplikacijų.
Daugelis inkstų ir kasos cistų yra nepiktybinės, tačiau turi būti nuolat stebimos ir šalinamos chirurgiškai, jei yra didesnės nei 3 cm (didesnė supiktybėjimo tikimybė).
Feochromocitoma yra retas antinksčių šerdinės dalies navikas, kuris gali lemti ypač aukštą kraujospūdį, todėl prieš kiekvieną chirurginę operaciją reikia tai atmesti (atliekami specialūs šlapimo katecholaminų tyrimai).
Ypač retai pasitaikantis endolimfatinio maišelio navikas (ELST) gali lemti klausos sumažėjimą,todėl reikia tikrinti klausą, esant jos silpnėjimo požymiams ir taikyti gydymą.
Kiti požymiai (sėklidės prielipo darinukai vyrams ir plačiojo gimdos raiščio darinukai moterims) nėra svarbūs kliniškai ir yra nepavojingi.
Iki 60 metų VHL geno mutaciją turintiems asmenims beiveik 100 proc. pasireikš vienoks ar kitoks požymis, todėl labai svarbu ilgalaikė tinkslinga stebėsena.
Požymių pasireiškimas yra labai individualus ir priklauso nuo genetinio pokyčio (mutacijos), todėl genetinis tyrimas yra labai svarbus ne tik patvirtinant ligą, bet ir ją prognozuojant.
Kaip paveldimas von Hippel-Lindau sindromas?
Von Hippel-Lindau ligą lemia 3-ioje chromosomoje esančio VHL geno mutacija (pakitimas), kuri dažniausiai būna paveldėta. Paveldėjimo tikimybė yra 50% (autosominis dominantinis paveldėjimas).
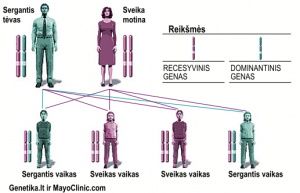
Autosominis dominantinis paveldėjimas
Autosominio dominantinio paveldėjimo atveju, pakitęs genas yra dominantinis, kas reiškia, kad pakanka tik vieno pakitusio geno, kad pasireikštų liga. Autosominiu dominantiniu būdu paveldima liga sergantis asmuo (šiame pavyzdyje tėvas), turi 50 proc. tikimybę, kad susilauks ta pačia liga sergantį vaiką (turintį pakitųsi dominantinį geną) ir 50 proc., kad susilauks sveiką vaiką (turintį du normalius genus). Šios tikimybės galioja kiekvienam nėšumui.
VHL geno produktas slopina perteklinį kraujagyslių ląstelių augimą, todėl sutrikus normaliai VHL geno funkcijai, dažniau atsiranda kraujagyslių „gumulėlių“ (angiomų arba hemangioblastomų). Genetinis VHL geno tyrimas leidžia nustatyti patogenines mutacijas daugeliui klinikinius požymius turintiems asmenims.
Onkogenetinis VHL geno tyrimas
Onkogenetinis VHL geno tyrimas yra vienintelis tikslios diagnozės būdas. VHL geno tyrimas atliekamas po gydytojo onkogenetiko konsultacijos. Konsultacijos metu bus patikslinta šeiminė ligos istorija (sudaroma genealogija), patikslintos diagnozės (todėl reikėtų turėti diagnozuotų būklių išrašus/kopijas), skiriamas onkogenetinis tyrimas, aptariama ligos eiga, gydymo ir prevencijos galimybės. Gydytojas onkogenetikas sudarys individualizuotą ligos stebėjimo planą ir padės jo laikytis.
Onkogenetinis pacientų tyrimas dėl VHL mutacijų turėtų būti atliekamas:
- bet kokio amžiaus asmenims su izoliuota tinklainės ar CNS hemangioblastoma (10-40% visų hemangioblastomų yra susijusios su VHL);
- bet kuriam asmeniui, kuriam diagnozuota feochromocitoma (neuroendokrininis antinksčių šerdies navikas) bet kuriame amžiuje, ypač jei yra abipusė arba daugiažidininė liga;
- bet kuris asmuo, kuriam diagnozuotas šviesių ląstelių inkstų vėžys (RCC) iki 40 metų, abipusis šviesių ląstelių inkstų vėžys (iki 60m.) arba yra stipri šeiminė šviesių ląstelių inkstų vėžio anamnezė.
- šeimoje nustatytas von Hippel-Lindau sindromas.
Dažnai, skiriant VHL geno tyrimą, tiklsinga atlikti šiuolaikinį kelių genų grupių tyrimą dėl paveldimo polinkio inkstų ir antinksčių navikams. Neradus genų pokyčių vaikams ar giminaičiams, šeimoje, kurioje nustatyta VHL geno mutacija, jiems (ir jų palikuonims) liga atmetama ir daugiau nereikia laikytis specialios patikros rekomendacijų.
Stebėsena esant von Hippel-Lindau sindromui
Pagrindinis VHL pacientų gydymo ir stebėsenos principas yra būdingų būklių ikisimptominis (t.y. dar iki joms pasireiškiant) nustatymas ir komplikacijų prevencija. Nustatyta, kad ankstyva diagnostika pagerina pacientų išgyvenamumą ir gyvenimo kokybę. VHL požymiai yra labai įvairūs ir skirtingi, netgi sergantiems toje pačioje šeimoje. Kadangi neįmanoma numatyti tikslios ligos pasireiškimo eigos (galima tik nustatyti tikimybę), reikalinga nuolatinė visų būdingų požymių patikra visą gyvenimą.
Tarptautinės (orientacinės) patikros ir stebėsenos rekomendacijos šeimoms su patvirtinta ar įtariama VHL liga:
| Amžius |
Dažnis |
Stebėsenos rekomendacijos |
| bet koks amžius | Šeimos su žinoma VHL geno mutacija turi būti informuotos apie ikisimptominio genetinio tyrimo galimybę;
prieš kiekvieną operaciją ar gimdymą reikia atmesti feochromocitomą. |
|
| nuo 5 metų | kas metus | Akių dugno tyrimas (oftalmologo konsultacijos);
Klinikinis ištyrimas (pagal būdingas organų sistemas, kraujospūdžio sekimas) |
| nuo 11-15 metų | kas metus | Vidaus organų (inkstų, kasos, antinksčių) ultragarsinis (UG, magnetinės rezonansinė tomografijos (MRT) arba kompiuterinės tomografijos (KT) tyrimas |
| nuo 15-16 metų | kas 2-3 metus | CNS kompiuterinė tomografija (KT) arba magnetinė rezonansinė tomografija (MRT) |
| nuo diagnozės | kas metus | gyd. onkogenetiko konsultacija |
P.S. Individualų stebėsenos planą ir rekomendacijas jums sudarys bei stebėjimą organizuos paveldimų onkologinių ligų specialistas gyd. onkogenetikas.
Daugiau informacijos (anglų k.)